Explore The Data
PROPEL: 52-week data
ERT-subgroup analysis: prespecified
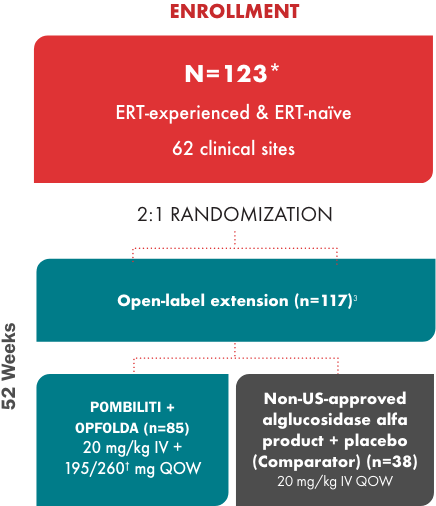
PROPEL study design1-3
Nearly 80% of the study population was ERT-experienced

- Primary endpoint: Change from baseline to Week 52 in 6-minute walk distance (6MWD) for comparison of superiority3‡
- Key secondary endpoint: Change from baseline to Week 52 in forced vital capacity (FVC)3
- POMBILITI in combination with OPFOLDA is not approved for use in ERT-naïve patients with LOPD. The ERT-naïve patient subgroup enrolled too few patients to conclusively interpret the data1,3
*After database lock, 1 ERT-naïve subject was found to have intentionally underperformed at baseline. This subject was excluded from all 6MWD efficacy results; this change did not alter the statistical outcomes of the study.4
†Dosage based on patient weight.2
‡Primary endpoint for superiority was not met.3
Baseline characteristics
Nearly 80% of the study population was ERT-experienced
Minimum treatment time for inclusion was 2 years


*After database lock, 1 ERT-naïve subject was found to have intentionally underperformed at baseline. This subject was excluded from all 6MWD efficacy results; this change did not alter the statistical outcomes of the study.4
†Dosage based on patient weight.2
‡Primary endpoint for superiority was not met.3
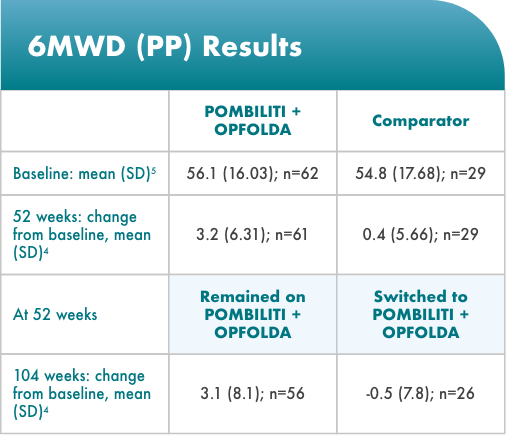
POMBILITI + OPFOLDA patients walked farther1,2*
6MWD mean change from baseline (± SE)
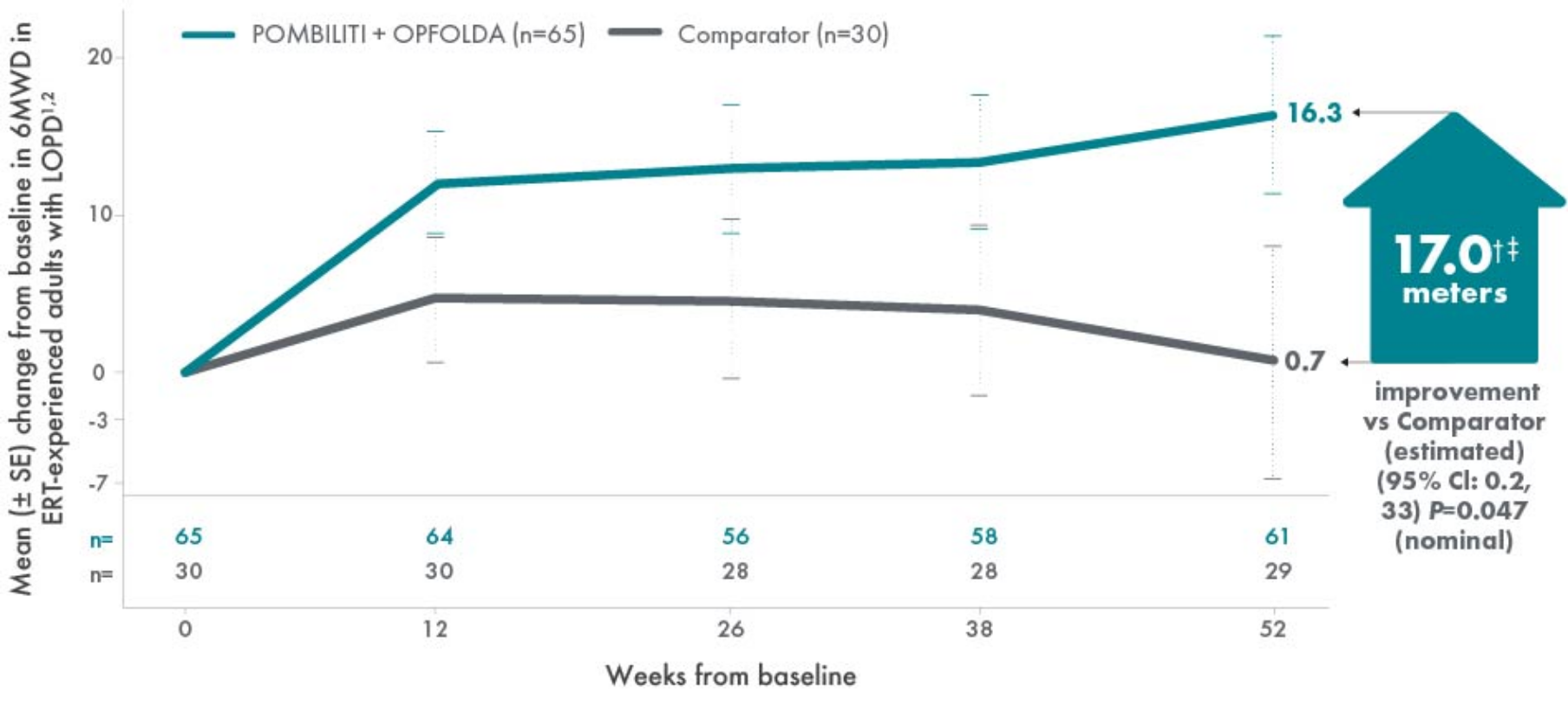
*The primary efficacy endpoint in PROPEL, change in 6MWD at 52 weeks in the POMBILITI + OPFOLDA arm versus the comparator arm, was not met.3
†A US-approved alglucosidase alfa product was not used in this clinical trial. Conclusions cannot be drawn from this clinical trial regarding comparative effectiveness between a US-approved alglucosidase alfa product and POMBILITI in combination with OPFOLDA.1,2
‡For the ERT-experienced group, the treatment difference of the mean was estimated by nonparametric analysis of covariance that included treatment, gender, baseline 6MWD, age, weight, and height in the model. Missing data at Week 52 was imputed using last observed values.1,2
POMBILITI + OPFOLDA patients’ lung function improved1,2*
Percent predicted (PP) forced vital capacity (FVC) in patients treated with POMBILITI + OPFOLDA1,2

*The primary efficacy endpoint in PROPEL, change in 6MWD at 52 weeks in the POMBILITI + OPFOLDA arm versus the comparator arm, was not met.3
†For the ERT-experienced group, the treatment difference of the mean was estimated by analysis of covariance that included treatment, gender, baseline FVC, age, weight, and height in the model. Missing data at Week 52 was imputed using last observed values.1,2
POMBILITI + OPFOLDA patients had improved biomarkers1,2
POMBILITI + OPFOLDA reduced Hex4 levels in ERT-experienced patients vs the comparator at 52 weeks1

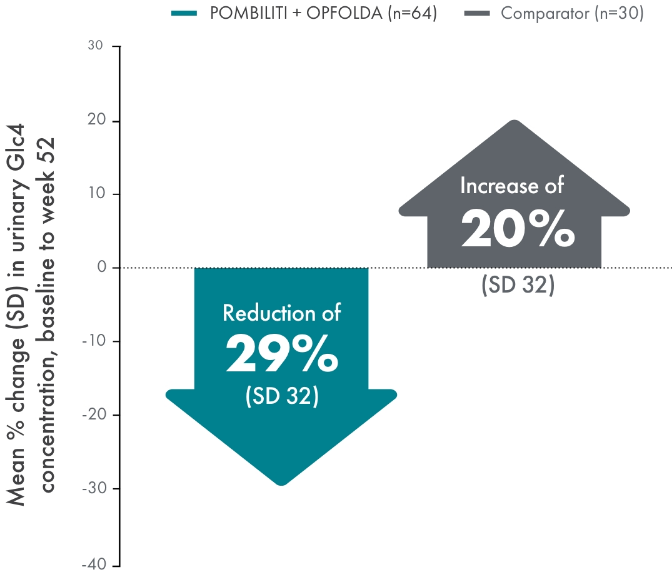
Baseline Glc4 concentration:
- POMBILITI + OPFOLDA: 4.6 mmol/mol
- Comparator: 7.2 mmol/mol
Hex4 is a metabolite of excess glycogen and is measured by Glc4 concentration.1
ERT-experienced population
Additional secondary endpoint scores in patients treated with POMBILITI + OPFOLDA (LS mean treatment difference from baseline)1,2

Demonstrated safety profile in 151 patients across 3 clinical trials1,2


Life-threatening hypersensitivity reactions, including anaphylaxis, have been reported in POMBILITI-treated patients1
- In clinical trials, 41 (27%) POMBILITI-treated patients experienced hypersensitivity reactions, including 4 (3%) reporting severe hypersensitivity reactions and 4 (3%) additional patients who experienced anaphylaxis1*
- Infusion-associated reactions were reported in 48 (32%) patients treated with POMBILITI + OPFOLDA across 3 clinical trials1
- There was no identified clinically significant effect of antidrug antibodies (ADAs) on POMBILITI + OPFOLDA PK or PD over the treatment duration of 52 weeks in PROPEL. Due to the small number of patients with negative ADA, the effect of ADA on the effectiveness of POMBILITI + OPFOLDA is unknown1
*Fulfilling at least one of the Sampson criteria.1
Open-label extension (OLE): 104-week data
ERT-subgroup analysis: prespecified from PROPEL
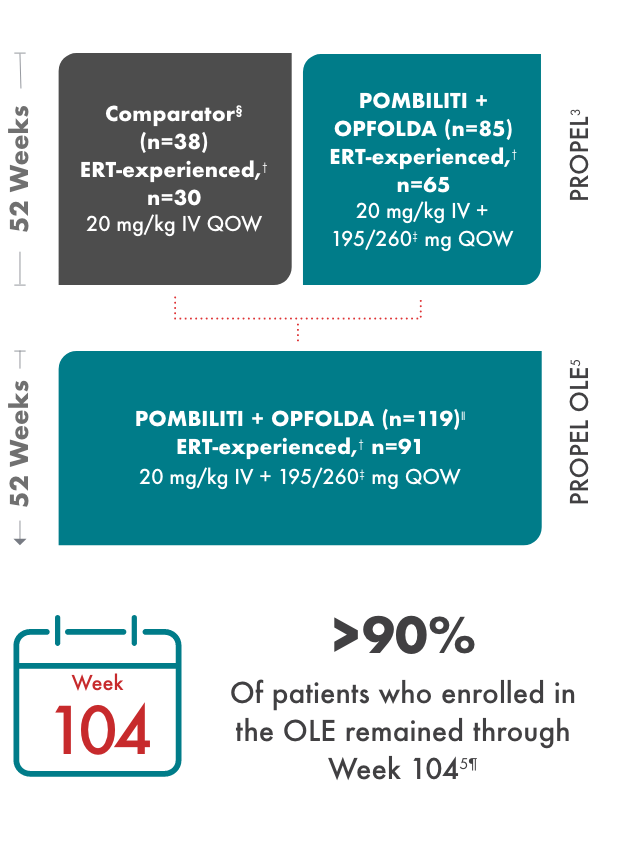
OLE study design
PROPEL Open-Label Extension (OLE): Interim analysis evaluating the long-term efficacy and safety of POMBILITI + OPFOLDA for an additional 52 weeks5
- After 52 weeks in the PROPEL study, 91 ERT-experienced patients and 28 ERT-naïve patients continued or crossed over to POMBILITI + OPFOLDA*†
- Patients in the OLE study received POMBILITI + OPFOLDA every 2 weeks, beginning 2 weeks after the last dose of the ERT that they received in the PROPEL study
- Baseline characteristics in PROPEL were representative of the LOPD population and generally similar between
treatment groups

Select Endpoints5
- Motor function: 6MWD (% predicted)
- Respiratory function: FVC (sitting, % predicted)
- Biomarkers: Hex4 (mmol/mol) and CK (U/L)
* POMBILITI in combination with OPFOLDA is not approved for use in ERT-naïve patients with LOPD. The ERT-naïve patient subgroup in PROPEL enrolled too few patients to conclusively interpret the data.1,3
† ERT‑experienced patients are defined as those treated with ERT for at least 2 years prior to their participation in the PROPEL study.3
‡ Dosage based on patient weight.1,2
§ A US-approved alglucosidase alfa product was not used in this clinical trial. Conclusions cannot be drawn from this clinical trial regarding comparative effectiveness between a US-approved alglucosidase alfa product and POMBILITI in combination with OPFOLDA.1,2
‖ Includes 1 patient who enrolled but was never dosed.5
¶ 11 patients discontinued due to withdrawn consent, adverse events, investigator decision, or because they were lost to follow-up.5
Important study limitations5
- The OLE was unblinded and had no control group
- This study includes data that are not in the FDA-approved Prescribing Informations for POMBILITI + OPFOLDA
- Data were analyzed descriptively, with no statistical comparisons. Therefore, no conclusions regarding treatment differences should be made as results could represent chance findings
- LOPD is a rare disease, so the sample size was relatively small
- Given the heterogeneous nature of LOPD, spanning a wide spectrum of manifestations, disease severity, rates of progression, and responses to treatment, tailoring the individualized treatment approaches to optimize patient outcomes will become more critical
- Creatine kinase (CK) is a nonspecific biomarker of muscle damage. The clinical relevancy of CK has not been established in LOPD6
6-Minute Walk Distance (6MWD)
Mean (± SE) change from PROPEL baseline in PP 6MWD in ERT-experienced adults with LOPD5
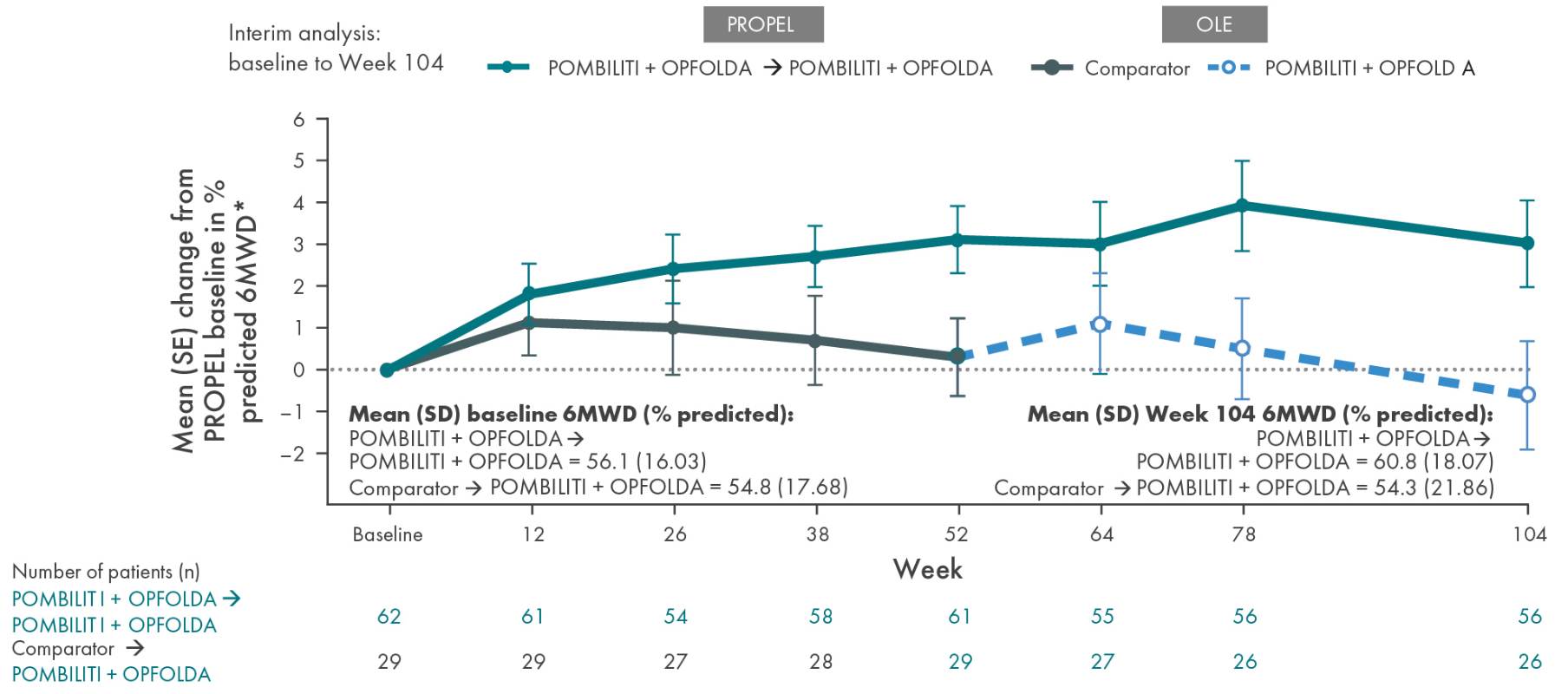
*The primary efficacy endpoint in PROPEL, change in 6MWD at 52 weeks in the POMBILITI + OPFOLDA arm versus the comparator arm, was not met.

Forced vital capacity (FVC)
Percent predicted (PP) FVC results over 104 weeks5



Biomarker: Hex4 levels
Hex4 is a metabolite of excess glycogen and is measured by Glc4 concentration. Elevated Hex4 levels may be indicative of Pompe disease1



Biomarker: serum creatine kinase (CK) levels
CK is a biomarker for muscle damage6*

*The clinical relevancy of changes in CK levels has not been established in LOPD.




Safety profile: no new safety signals were identified during the OLE3
- Most TEAEs were mild to moderate in severity
- The most common TEAEs were headache, diarrhea, pyrexia, fatigue, and nausea
- Five patients withdrew from the study due to TEAEs experienced during the OLE


*Includes data from patients treated with POMBILITI + OPFOLDA in PROPEL who may or may not have continued POMBILITI + OPFOLDA in the OLE, including data from both PROPEL and the OLE.4
†Includes data from the OLE only.5

Sign up for emails
Sign up to receive updates about LOPD and POMBILITI + OPFOLDA
6MWD, 6-minute walk distance; Alg, alglucosidase alfa; CI, confidence interval; Cipa, cipaglucosidase alfa; CK, creatine kinase; ERT, enzyme replacement therapy; FVC, forced vital capacity; Glc4, glucosidase tetrasaccharide; GSGC, Gait, Stairs, Gowers’ maneuver, Chair; Hex4, hexose tetrasaccharide; IARs, infusion-associated reactions; IU, international units; IV, intravenous; LOPD, late-onset Pompe disease; LS, least squares; MEP, maximal expiratory pressure; Mig, miglustat; MIP, maximal inspiratory pressure; MMT, manual muscle test; OLE, open-label extension; PD, pharmacodynamics; PK, pharmacokinetics; PP, percent predicted; PROMIS, Patient-Reported Outcomes Measurement Information System; QOW, every other week; SD, standard deviation; SNIP, sniff nasal inspiratory pressure; TEAEs, treatment-emergent adverse events.